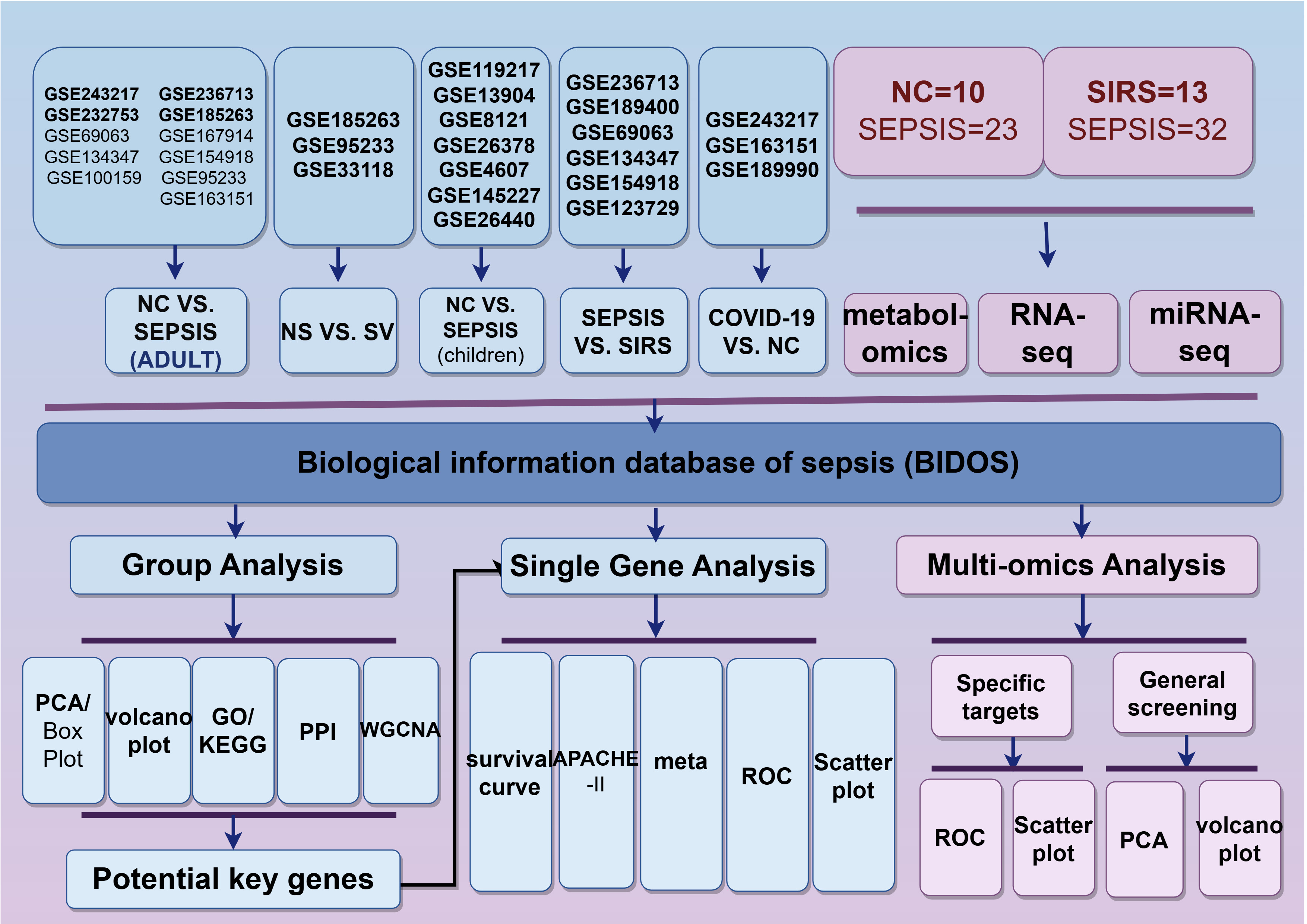
Welcome to the Bioinformatics Database Of Sepsis (BIDOS)! This platform is dedicated to the comprehensive study of multiple omics data from public datasets, exploring the potential pathogenesis of sepsis and screening potential core targets. This study collected a total of 30 high-quality public datasets based on SEPSIS 3.0 standards for adults after 2016, as well as datasets for children with septic shock. Among them, GSE65682 includes 28 day prognostic data of over 400 sepsis patients; GSE54514 contains APACHE II scoring data. This platform is divided into three modules: single gene analysis, group analysis, and multi omics analysis. 1) Single gene analysis,is mainly used to explore the relationship between specific genes and the prognosis of sepsis patients as well as APACHE II; Meta analysis of large samples was conducted to explore the differences in the expression of specific genes in different groups: sepsis and normal group (ADULT), sepsis and normal group (children), sepsis and systemic inflammatory response syndrome (SIRS), survival (SV) and non-survival group (NS), COVID-19 patients (COVID-19) and normal group (NC). 2) Group analysis, bioinformatics analysis of selected public datasets, potential targets can be preliminarily screened through PPI network and WGCNA co expression analysis. 3)Multiomics analysis, this study included plasma metabolomics and transcriptomics data from sepsis (n=55), normal group (n=10), and SIRS (n=13) patients conducted by our team. Researchers can conduct subsequent studies using both conventional screening and specific target input methods. The trial received approval from the Ethics Committee of the Afliated Hospital of Southwest Medical University (Ethics No.ky2018029), Clinical Trial Registration No. Te study ChiCTR1900021261 adheres to the principles of the Declaration of Helsinki. The sequence data analyzed in this research is available in the Chinese National Genebank database (CNGBdb) and can be accessed at https://db.cngb.org/. The accession number is CNP0002611.
- Single Gene Analysis
- Group Analysis
- Multi-omics Analysis
Melioidosis, caused by Gram negative bacteria Burkholderia pseudomallei, is a major type of community-acquired septicemia in Southeast Asia and Northern Australia with high mortality and morbidity rate. More accurate and rapid diagnosis is needed for improving the management of septicemic melioidosis. We previously identified 37-gene candidate signature to distinguish septicemic melioidosis from sepsis due to other pathogens. The aims of this current study were to independently validate our previous biomarker and consolidate gene selection from each of our microarray data set for establishing a targeted assay
The study included seventy-two critically ill patients admitted to the intensive care unit (ICU) of Nepean Hospital, Sydney, Australia. Of these, fifty-five patients were diagnosed to have sepsis, as confirmed by microbiological culture. The remaining seventeen patients did not have sepsis and were therefore used as controls. The study was approved by the hospital ethics committee and informed consent was obtained from all patients or their relatives.
159 consecutive traumatized patients were included in the study upon admission to the intensive care unit (ICU), fulfilling all of the following inclusion criteria: severe injuries to at least two body regions or three major fractures, between 18 and 65 years of age, an estimated Injury Severity Score (ISS) of ≥15 points after thorough assessment of injuries, <12 hours between the occurrence of the accident and the time of admission to the ICU, and at least greater than 3 days of survival. None of the patients underwent neuro- or cardiac surgery. We excluded patients with severe intracranial head injuries, coagulation abnormalities known at the day of admission to the ICU, acute renal failure, liver failure, malignant disease, or hemofiltration in the patient`s history. The Patients were divided into two groups depending on their posttraumatic course: Patients not complicatd by sepsis (Group: N) and Patients complicated by sepsis (Group: S). Using the CodeLink UniSet Human 10 K Bioarrays (GE Healthcare, Freiburg, Germany), we searched for early differences in the transcriptome of these two groups. Only whole blood samples drawn upon admission to the ICU were used for this investigation. Microarray results were validated by quantification of mRNA by TaqMan® technology. Each patient sample was hybridized in duplicate or triplicate on microarrays (label-extract technical replicate). A total of 72 arrays (36 group N, 36 group S) were subjected to microarray quality analysis. Of these, 2 arrays (0 group N, 2 group S) were excluded from the data set due to quality reasons and a final set of 70 arrays (36 group N and 34 group S) were subjected to microarray analysis. Since the eliminated 2 arrays belonged to patients with three replicates, they were treated as two replicates in the further analysis. The arrays were designated N_xx_yy_z or S_xx_yy_z, with x for the patient-ID (1,2,3,4..) and z for the technical replicate number (1,2 or 3).
There is currently no reliable tool available to measure immune dysfunction in septic patients in the clinical setting. This proof-of-concept study assesses the potential of gene expression profiling of whole blood as a tool to monitor immune dysfunction in critically ill septic patients. Whole blood samples were collected daily for up to 5 days from patients admitted to the intensive care unit with sepsis.Daily PAXgene samples for up to 5 days for sepsis survivors (n=26), sepsis nonsurvivors (n=9), and healthy controls (n=18).
This study sequenced peripheral blood RNA of 129 representative subjects with systemic inflammatory response syndrome (SIRS, n=23) or sepsis (infection with SIRS), including 78 sepsis survivors and 28 sepsis nonsurvivors, who had previously undergone plasma proteomic and metabolomic profiling.
Transcriptional profiling of granulocytes from 24 subjects was used to investigate their differences regarding sepsis and aging.Two-colour hybridization design. Aliquots of total RNA isolated from adult or elder individuals with sepsis or healthy controls were labeled with Cy3 and co-hybridized with a common reference RNA pool (Universal Human Reference RNA, Agilent, cat #740000) labeled with Cy5 . Biological replicates:6 per group. No technical replicates.
Total RNA from whole blood obtained from patients with sepsis caused by B.pseudomallei (n=29) or other pathogens (n=54) and uninfected controls (28 healthy and 27 subjects with type 2 diabetes mellitus) were collected. In order to validate the published signature, microarray data were generated from these samples. This dataset was also used for an independent selection of signature for septicemic melioidosis. The same RNA samples were used for validation by a high throughput real-time PCR technique, Fluidigm.
Retrospective, mutli-site sutdy using retrospective physician adjudication as a comparator.Systemic inflammation is a whole body reaction that can have an infection-positive (i.e. sepsis) or infection-negative origin. It is important to distinguish between septic and non-septic presentations early and reliably, because this has significant therapeutic implications for critically ill patients. We hypothesized that a molecular classifier based on a small number of RNAs expressed in peripheral blood could be discovered that would: 1) determine which patients with systemic inflammation had sepsis; 2) be robust across independent patient cohorts; 3) be insensitive to disease severity; and 4) provide diagnostic utility. The overall goal of this study was to identify and validate such a molecular classifier. Methods and Findings: We conducted an observational, non-interventional study of adult patients recruited from tertiary intensive care units (ICU). Biomarker discovery was conducted with an Australian cohort (n = 105) consisting of sepsis patients and post -surgical patients with infection-negative systemic inflammation.
51 septic shock patients and 22 healthy volunteers were included in this study. Septic shock patients were sampled twice, at admission, and a second time at D2 or D3. Admission samples from septic shock patients were compared to healthy volunteers, and according to day 28 survival status. Modulation of gene expression between the 2 time points was also analyzed according to day 28 survival.
Development of a cost-effective and practical targeted transcriptional fingerprinting assay (TFA) for monitoring a diverse repertoire of immune responses identified in the human blood transcriptome relying on a modular transcriptional repertoire built on the basis of variations in transcript abundance measured on a genome wide scale in the blood of nearly 1000 subjects across 16 immunologic conditions.
Genome-wide gene expression profiling of whole blood leukocytes in critically ill patients with sepsis or non-infectious disease has been used extensively in search of diagnostic biomarkers, as well as prognostic signatures reflecting diseases severity and outcome. Through technological advances in genomics it has become clear that transcription is not limited to protein-coding regions of the genome.
Gene expression changes in the blood was studied by RNAseq Results: Strong differences between patients groups, specific expression changes for heathy control (Hlty), uncomplicated infection (Inf1_P), sepsis (Seps_P), septic shock (Shock_P), follow-up of sepsis (Seps_FU), follow-up of septic shock (Shock_FU) groups.
To elucidate key pathways in the host transcriptome of patients infected with SARS-CoV-2, we used RNA sequencing (RNA Seq) to analyze nasopharyngeal (NP) swab and whole blood (WB) samples from 333 COVID-19 patients and controls, including patients with other viral and bacterial infections. Analyses of differentially expressed genes (DEGs) and pathways was performed relative to other infections
Identifying at first clinical presentation gene expression signatures that predict subsequent severity will allow clinicians to identify the most at risk groups of patients, and also enable appropriate antibiotic use. Accordingly, we characterized the blood immune profiles of patients with early/pre-sepsis to identify signatures reflecting disease severity, organ dysfunction, mortality, and specific endotypes/mechanisms
This study aimed to compare gene expression profiles between patients with sepsis and healthy volunteers, to determine the accuracy of these profiles in diagnosing sepsis, and to predict sepsis outcomes by combining bioinformatics data with molecular experiments and clinical information.
Early diagnosis of sepsis and discrimination from SIRS is crucial for clinicians to provide appropriate care, management and treatment to critically ill patients. Here, we describe identification of transcriptional mRNA biomarkers able to identify severe systemic inflammation and differentiate Sepsis from SIRS, in adult patients within a multi-center clinical study. All patients were recruited in Intensive Care Units (ICUs) from multiple UK hospitals including 59 patients with abdominal sepsis, 84 patients with pulmonary sepsis, 42 SIRS patients with Out-of-Hospital Cardiac Arrest (OOHCA), at four time points including 30 healthy control donors
This prospective observational study conducted at Osaka University Graduate School of Medicine aimed to compare host responses in sepsis and COVID-19 patients by analyzing mRNA and miRNA profiles. They included 22 sepsis patients, 35 COVID-19 patients, and 15 healthy subjects. Sepsis was diagnosed using Sepsis-3 criteria, while COVID-19 was confirmed through SARS-CoV-2 RT-PCR testing and chest CT scans for pneumonia assessment
Gene expression was profiled in peripheral blood samples collected from patients during anaphylaxis, trauma, or sepsis, and from healthy controls. Patients were recruited from three Australian emergency departments (ED) between March 2011 and June 2013. Samples were collected at ED arrival (T0), 1 hour later (T1), and 3 hours post arrival (T2).
Timely and reliable distinction of non-infectious systemic inflammatory response syndrome (SIRS), common in critically ill patients, from sepsis to support adequate antimicrobial therapy safes lives but is clinically challenging. Expeditious sepsis biomarkers are thus urgently sought. Blood transcriptional profiling provides insights into sepsis pathophysiology, but variability in leukocyte subtype composition complicates profile interpretation, and reliable reference genes to normalize gene expression in sepsis are lacking.
Here we developed a new approach to sepsis diagnosis that integrates host transcriptional profiling with metagenomic broad-range pathogen detection from cell-free plasma RNA and DNA.
Septic shock by pneumopathy was studied in prospective way in 20 patients and compared to normal blood. Blood samples were taken within 12 hours of diagnosis and the resulting transcritomes were compared to 42 normal control samples (selected samples from series GSE10715, GSE12711, GSE16728 and GSE7400). keywords: patient cohort study, septic shock, transcription-profile, blood
Infection with the SARS-CoV2 virus can vary from asymptomatic, flu-like with moderate disease, to critically severe. Severe disease, termed COVID-19, involves acute respiratory deterioration that is frequently fatal. To understand the highly variable presentation, and identify biomarkers for disease severity, blood RNA from COVID-19 patient in an intensive care unit was analyzed by whole transcriptome RNA sequencing. Both SARS-CoV2 infection and the severity of COVID-19 syndrome were associated with up to 25-fold increased expression of neutrophil-related transcripts, such as neutrophil defensin 1 (DEFA1), and 3-5-fold reductions in T cell related transcripts such as the T cell receptor (TCR).
Around 42,000 children suffer from severe sepsis each year in the US alone, resulting in significant morbidity, mortality and billion dollar expenditures in the US healthcare system. Sepsis recognition is a clinical challenge in children. Biomarkers are needed to tailor appropriate antimicrobial therapies and improve risk stratification. The goal of this study was to determine if gene expression profiles from peripheral blood were associated with pathogen type and sepsis severity in children treated for suspected sepsis.
Normal children, children with SIRS, children with sepsis, and children with septic shock. Objectives: To advance our biological understanding of pediatric septic shock, we measured the genome-level expression profiles of critically ill children representing the systemic inflammatory response syndrome (SIRS), sepsis, and septic shock spectrum.
Sepsis represents a complex disease with dysregulated inflammatory response and high mortality rate. Long noncoding RNAs (lncRNAs) have been reported to play regulatory roles in a variety of biological processes. However, studies evaluating the function of lncRNAs in pediatric sepsis are scarce, and current knowledge of the role of lncRNAs in pediatric sepsis is still limited. We explored the expression patterns of both lncRNAs and mRNAs between pediatric sepsis patients and healthy controls based on a comprehensive microarray analysis.
Background: Septic shock heterogeneity has important implications for the conduct of clinical trials and individual patient management. We previously addressed this heterogeneity by indentifying 3 putative subclasses of children with septic shock based on a 100-gene expression signature corresponding to adaptive immunity and glucocorticoid receptor signaling. Herein we attempted to prospectively validate the existence of these gene expression-based subclasses in a validation cohort.
Background: Septic shock is a heterogeneous syndrome within which probably exist several biological subclasses. Discovery and identification of septic shock subclasses could provide the foundation for the design of more specifically targeted therapies. Herein we tested the hypothesis that pediatric septic shock subclasses can be discovered through genome-wide expression profiling. Methods: Genome-wide expression profiling was conducted using whole blood-derived RNA from 98 children with septic shock, followed by a series of bioinformatic approaches targeted at subclass discovery and characterization.
Goal of the experiment: To identify correlated genes, pathways and groups of patients with systemic inflammatory response syndrome and septic shock that is indicative of biologically important processes active in these patients. Background: We measured gene expression levels and profiles of children with systemic inflammatory response syndrome (SIRS) and septic shock as a means for discovering patient sub-groups and gene signatures that are active in disease-affected individuals and potentially in patients with poor outcomes.
In an ongoing translational research program involving microarray-based expression profiles in pediatric septic shock, we have now conducted longitudinal studies focused on the temporal expression profiles of canonical signaling pathways and gene networks. Genome-level expression profiles were generated from whole blood-derived RNA samples of children with septic shock (n = 30 individual patients) corresponding to days 1 and 3 of admission to the pediatric intensive care unit.